Болести на опорно двигателната система, Здравето от А до Я
ОСТЕОГЕНЕЗА ИМПЕРФЕКТА – OSTEOGENESIS IMPERFECTA
Това ще ви помогне
Специално подбрани от нас продукти, помагащи при описаните в статията здравословни проблеми.




Генетично заболяване, при което костите са крехки и лесно се получават фрактури.
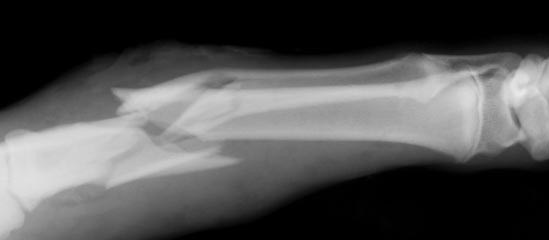 Osteogenesis imperfecta (ОИ) засяга приблизително 1 на всеки 20 000 души, като един на всеки 50 000-60 000 страда от много тежка форма на заболяването. Болните имат нарушение в синтеза на колаген (здравия белтък, влизащ в състава на костите, хрущялите и съединителната тъкан): организмът им или не произвежда достатъчно колаген, или произведеният е дефектен. Това прави костите крехки и лесночупливи.
Osteogenesis imperfecta (ОИ) засяга приблизително 1 на всеки 20 000 души, като един на всеки 50 000-60 000 страда от много тежка форма на заболяването. Болните имат нарушение в синтеза на колаген (здравия белтък, влизащ в състава на костите, хрущялите и съединителната тъкан): организмът им или не произвежда достатъчно колаген, или произведеният е дефектен. Това прави костите крехки и лесночупливи.
Кои са причините?
Заболяването се развива в резултат на генетичен дефект, но понякога може да се появи и спонтанно. Тъй като всеки човек притежава по две копия от съответния ген, вероятността детето на човек, страдащ от заболяването, да наследи един нормален и един дефектен ген (съответно да наследи и заболяването) е 50%. Съществуват четири известни форми на ОИ:
Тип I е най-честата, но и най-леката форма на заболяването, която обикновено се предава по наследство. Тя се дължи на намалено количество на синтезирания колаген. Повечето фрактури се получават преди настъпването на пубертета. Освен крехки кости при хората с ОИ тип I често се наблюдават и синьо оцветяване на бялото на очите, гръбначни изкривявания, тънка кожа, халтави стави и ронливи зъби. Във възрастта между 20 и 30 години могат да се развият нарушения на слуха.
Тип II засяга около 10% от общия брой пациенти и представлява най-тежката форма на заболяването. Той се дължи на спонтанна генетична мутация, определяща дефектна структура на колагена, която причинява чупливост и деформации на костите. Това води до дихателни нарушения, които обикновено завършват фатално скоро след раждането.
Тип III също е резултат от спонтанна мутация, определяща дефектна структура на колагена, но не протича толкова тежко, колкото тип II. Повечето от фрактурите се наблюдават главно преди пубертета, като могат да се получат и по време на вътреутробния живот на плода. Пациентите притежават повечето от белезите, характерни за тип I, като освен тях могат да имат нисък ръст, огъващи се кости и костни деформации. Понякога се наблюдават и проблеми с дишането.
Тип IV обикновено се предава по наследство, въпреки че понякога може да се развие и в резултат от спонтанна мутация. Той протича по-леко от тип III, но е по-тежък от тип I. При него фрактурите също са по-чести преди пубертета и пациентите имат нисък ръст.
Диагноза и методи на лечение
Лекарят може да постави първоначална диагноза по данните от прегледа. Потвърждаващите я изследвания включват изследване на структурата на колагена и генетичен анализ.
Понастоящем заболяването е нелечимо, така че терапията цели да облекчи симптомите и да поддържа костите и мускулите колкото е възможно по-здрави. За облекчение на болките и увеличаване на мускулната сила се препоръчва физиотерапия. Плуването укрепва костите и мускулите без опасност от фрактури; ходенето също е подходящо упражнение. Здравословната диета, богата на калций и витамин D, е важна за здравината на костите.
Болката се контролира с помощта на студени и топли компреси, медикаменти и понякога нервна блокада, при която болкоуспокояващи лекарства се инжектират около засегнатия нерв. Получените фрактури трябва да бъдат лекувани с повишено внимание, като в по-тежките случаи в дългите кости се въвеждат метални пръчки, които поддържат здравината им и ги предпазват от деформиране.
Изгледите за изхода от болестта са много различни в зависимост от общото състояние на пациента и типа на ОИ. Докато новородените с ОИ тип II рядко оцеляват дълго след раждането, при някои хора с тип I симптомите на заболяването са толкова леки, че то никога не бива диагностицирано.
Материалите в lekuva.net са авторски и може да се използват само с публикуване на активен dofollow линк към оригиналния текст и без промяна на съдържанието, запазвайки всички линкове!
Това ще ви помогне
Специално подбрани от нас продукти, помагащи при описаните в статията здравословни проблеми.